


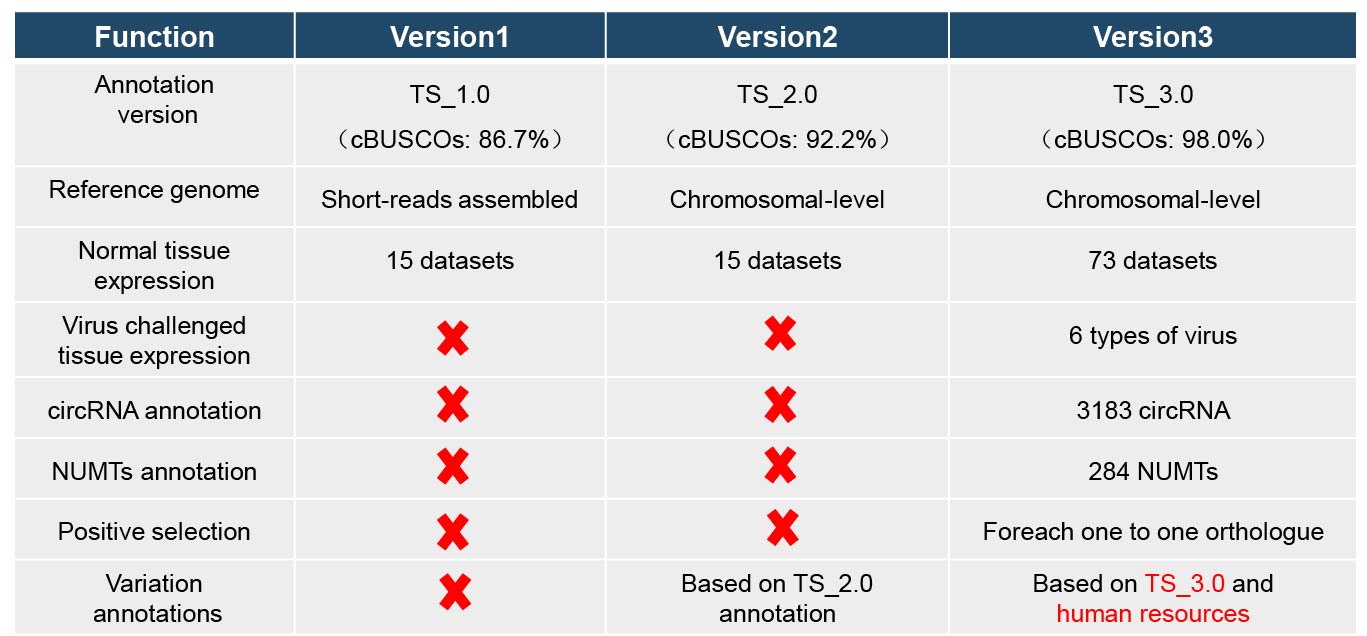
Treeshrew Database is an integrated database for genome biology of treeshrew. This database provides not only information of genomic data including sequences, genes and functional annotation, but also biological information such as expression data and corresponding references. In addition, previously and newly developed bioinformatics tools provided in the database will facilitate users to easily exploit information they wanted. The first version of the Treeshrew Database version 1.0 was published on 2015, with the first high quality tree shrew reference genome (Fan 2012, Nature communications). The second version of TreeshrewDB, version 2.0, was released in 2019 with chromosomal level tree shrew reference genome. With the continuing production of tree-shrew-related omics data, the demands for retrieving, analyzing, and visualizing these up-to-date data have been emerging.
Here, we announced an updated version of TreeshrewDB (TreeshrewDB_3.0). In this version, we provided the following update: 1) Multiple ways to retrieve the most recent tree shrew genome annotations and newly annotated non-coding elements (circle RNA and nuclear mitochondrial DNA segments); 2) Substantially expanded the transcriptomic datasets collection, provided the expression data of 76 normal tissues and 6 types of viral challenged data retrieval function; 3) More comprehensive annotations of Chinese tree shrew genome variations based on the latest tree shrew genome annotations and human variation annotation resources. The comprehensively updates of the tree shrew genome database were aimed to provide up-to-date and user-friendly accesses to tree shrew genomic and transcriptomic data.
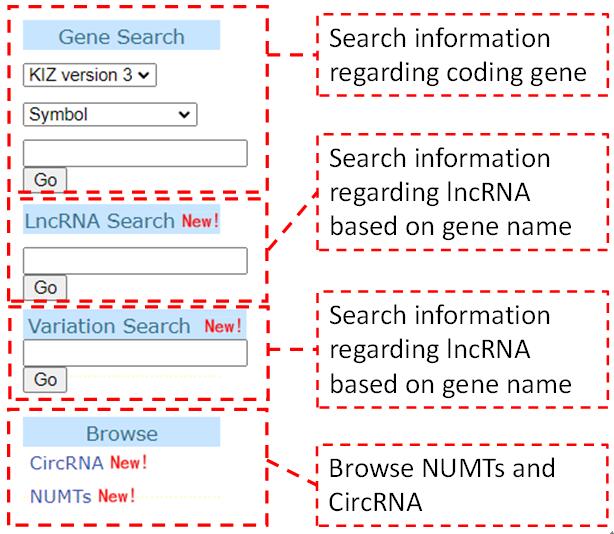
The quick search tool that users get gene and non-coding elements information quickly through gene symbol on the home page. We provide quick search for coding RNA, Long-non-coding RNA, genomic variation, circRNA and NUMTs.
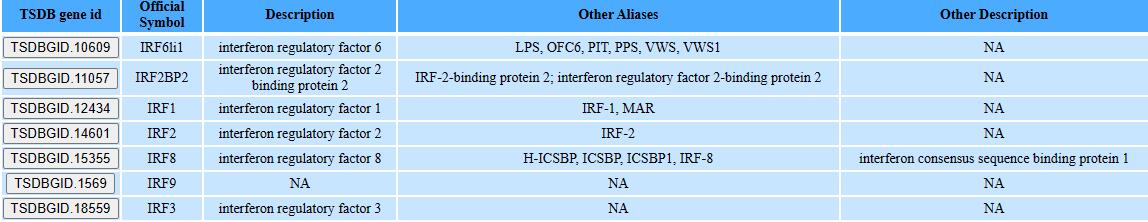
On the search page, we provided a “one-stop” retrieval system for viewing gene information by gene symbol, gene ID or gene full name.
Step1: Type the gene symbol, gene ID or gene full name

Step2: select the gene you want by click the TSDB gene id
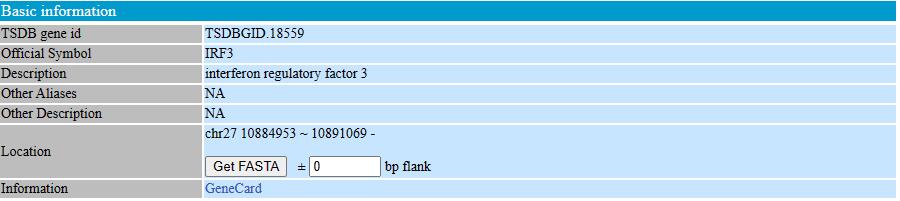
Step3: View the gene information
In the basic information table, users would view some basic information of the gene they want, including gene symbol, refseq ID, location, genomic map, or more from other databases. In the location region, users would obtain the genomic DNA sequence or with the flanking sequence of the gene they queried.
The sequences table is linked to the corresponding CDS sequence, UTR sequence, exon and intron sequence, as well as deduced protein sequence.

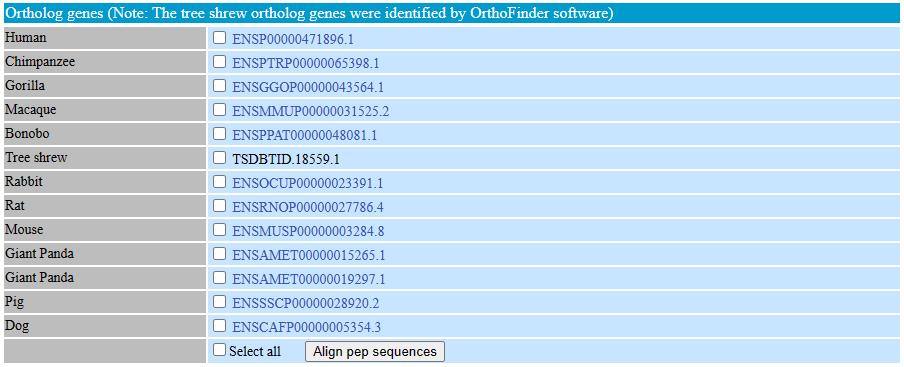
The ortholog table displayed all identified ortholog genes with links to the Ensembl database (http://www.ensembl.org). In order to facilitate to compare the ortholog gene sequences in different species, we provided a button which can quick align the selected sequences.
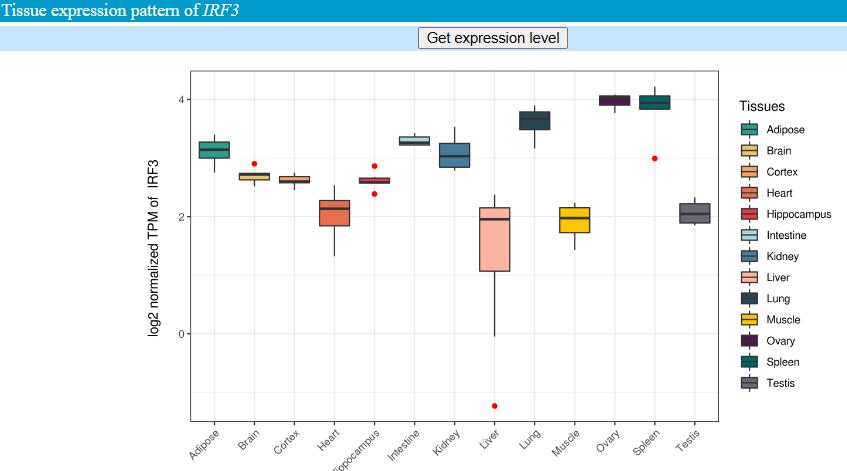
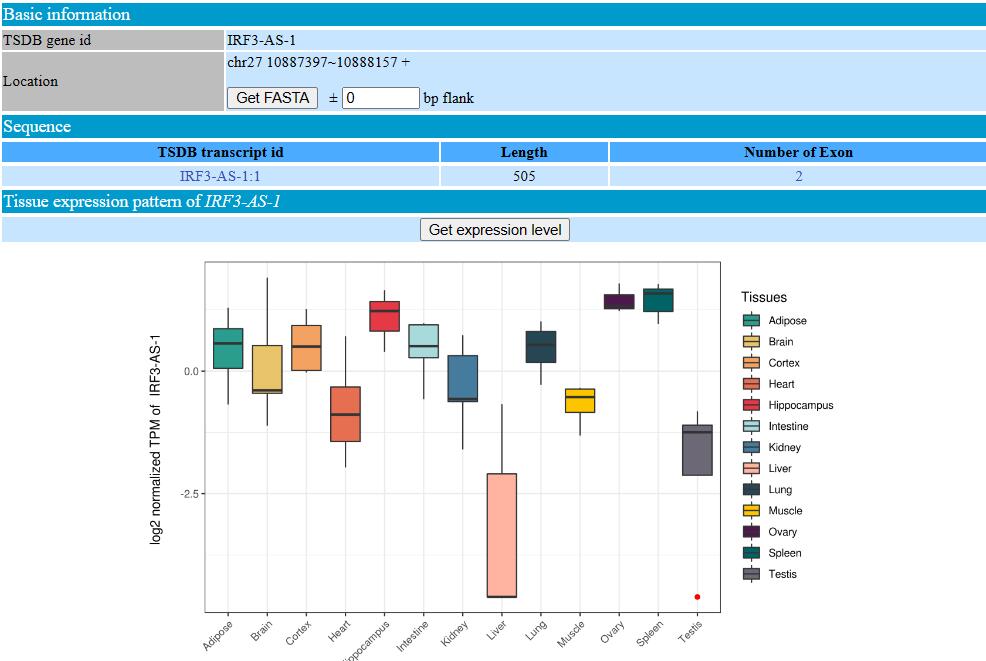
In the “tissue expression pattern” sector, we provided expression level of 13 tissues/organs, contain a total of 76 transcriptomic datasets. The original TPM level can be extracted by click “Get expression level” button. For visualization, we generated violin plot based on log2 normalized TPM.
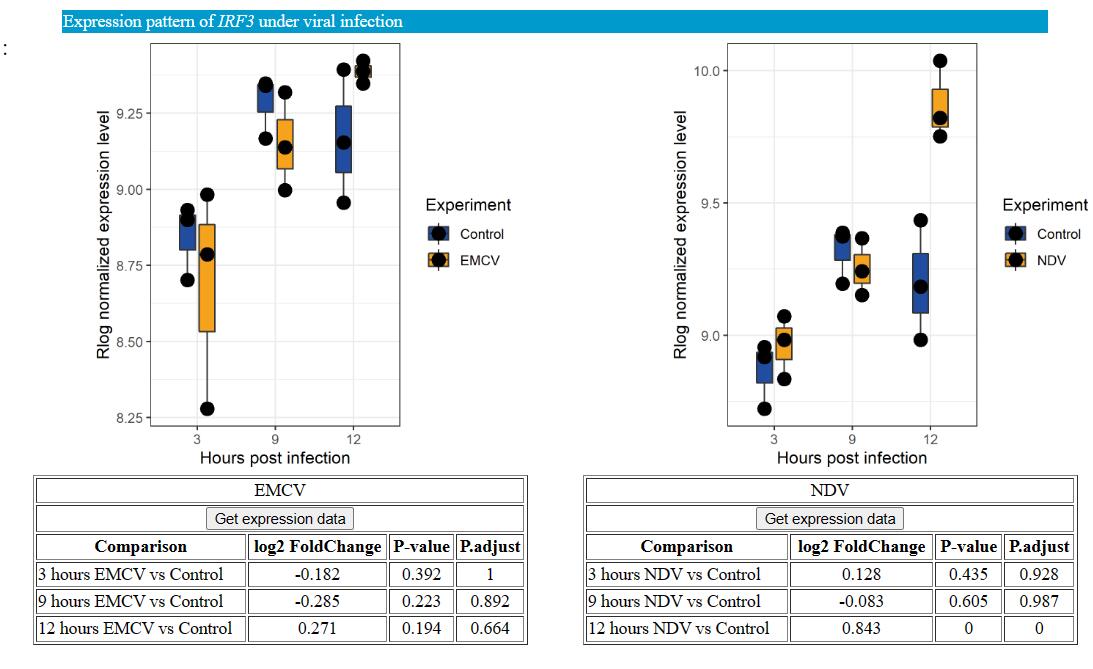
In the “expression under viral infection” sector, we provide expression level of tissue/cells infectious with 6 types of viruses. Including DNA virus herpes simplex virus type 1 (HSV-1), hepatitis B virus (HBV), RNA viruses Sendai virus (SeV), encephalomyocarditis virus (EMCV), Newcastle disease virus (NDV), and Zika virus (ZIKV). In brief, the host of HSV-1, EMCV, NDV, and SeV virus were tree shrew primary renal cell. The host of HBV was tree shrew liver, and the host of ZIKA was tree shrew peripheral blood mononuclear cell. In addition to baseline expression, expressional changes under virus infection for each gene were also provided. The rLog and TPM normalized expression, and results of differential expression (The log2 foldchange, P value, and adjusted P values) for each gene under viral infection can also be retrieved.
In the “variation” sector, we provided more comprehensive function annotations for genomic variations of wild tree shrew population and tree shrew inbreeding lines, including single nucleotide variations (SNVs) and sort insertion/deletion (InDel). In order to get a glimpse into tree shrew population genetics, we included whole genome sequencing data of 61 tree shrews in this study, including 7 wild tree shrew and 54 individuals from tree shrew inbreeding lines. We provided annotation information and allele frequencies (both wild tree shrew and all tree shrew populations) for variations located within 5000 bp upstream/downstream of each coding gene.

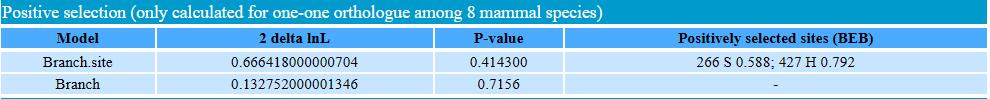
In the “positive selection” sector, we provided results of positive selection analysis for one to one orthologous (identified by OrthoFinder software) among 8 species, including human, gorilla, chimpanzee, macaca, tree shrew, mouse, rat, and dog. We use PAML software to identify positive selection signal using branch model and branch-site model.
In the “population genetics” sector, we provided detailed information regarding tree shrew genetics features. For every coding gene, we calculated their population genetic parameters based on variations located on their respective locus. The parameters including Tajima's Pi (θπ), Tajima's D, Watterson theta estimate (θw), Fu & Li's D, Fu & Li's F, and Fay & Wu's H.

In the function annotation table, the gene function annotation is displayed. Users would learn gene function classification from GO annotation, pathway information from KEGG annotation.

The lncRNA retrieval is provided to view the basic information and expression patterns regarding lncRNA of Chinese tree shrew.
Step1: Type the lncRNA name, or coding gene name

Step2: Select the ncRNA you want
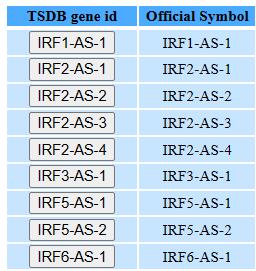
Step3: View the basic lncRNA information and expression pattern.
The detailed information is similar with coding gene.
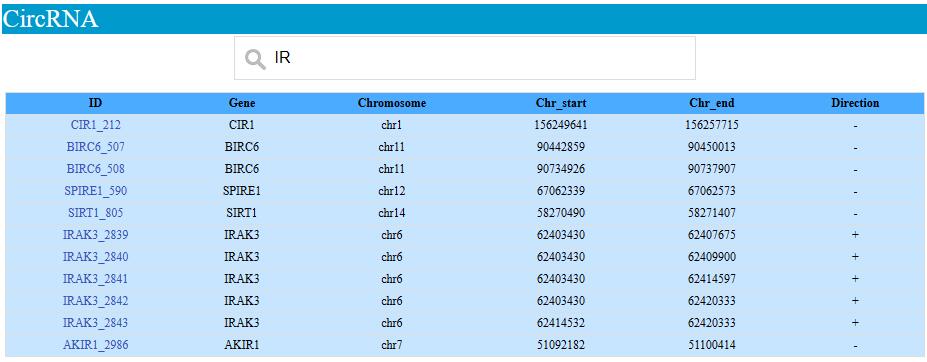
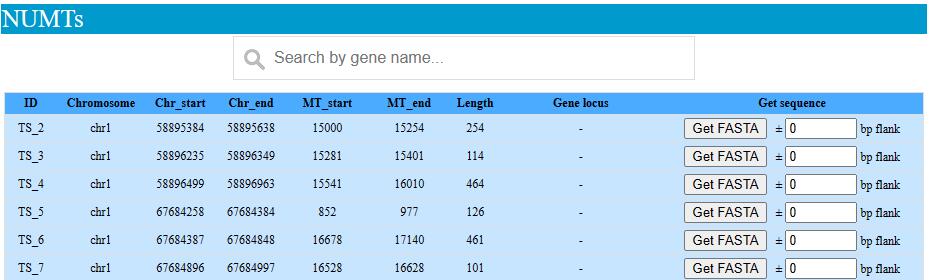
We provide browse and search function for tree shrew NUMTs and circRNA. NUMTs and circRNA can be searched by the coding gene locus they located on.
circRNA sequnce can be retrived by click the circRNA ID (first colum of the table)
NUMTs sequnce can be retrived by click the Get FASTA botton
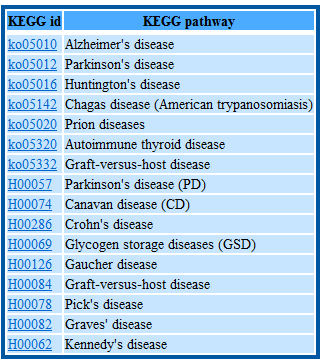
Users would search genes by GO term, domain information and pathway information taxonomy and download gene sequences by function classification in batch.
Step1: Tpye the KEGG pathway name

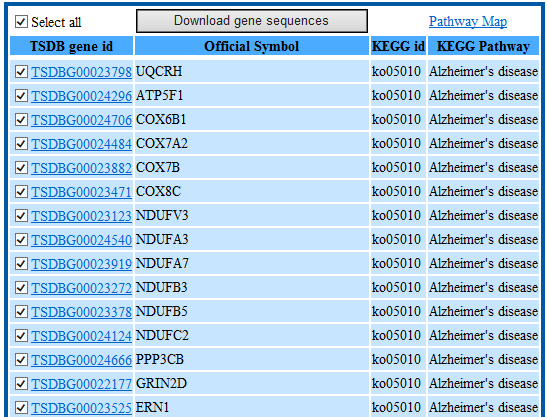
Step2: Select the pathway you want
Step3: View the genes information or select the genes you want to download the CDS and/or protein sequences in batch
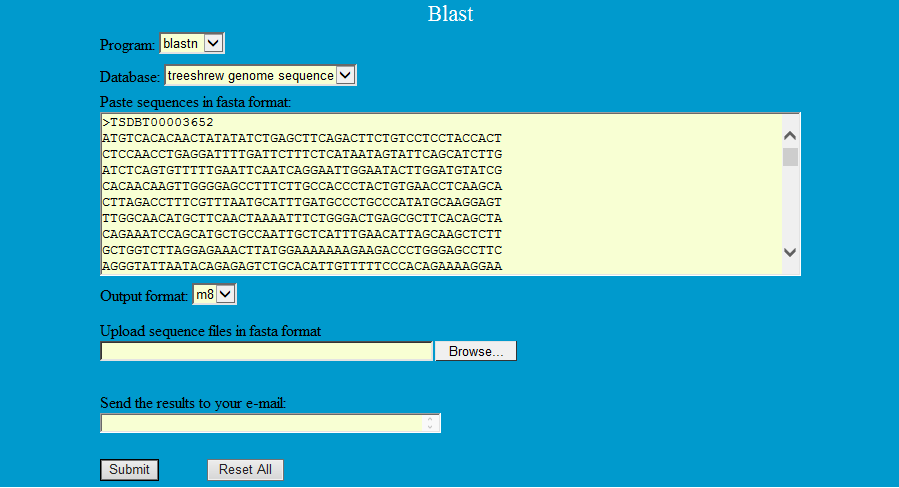
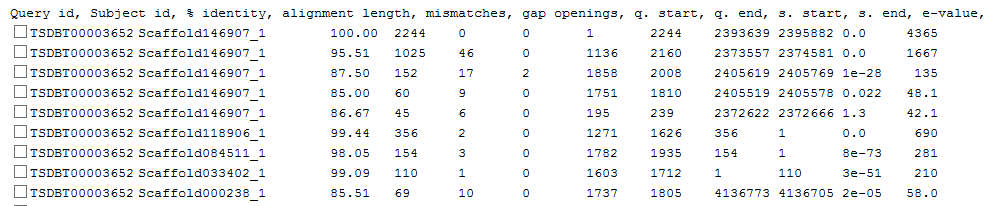
The Basic Local Alignment Search Tool (BLAST) finds regions of local similarity between sequences (Altschul et al. 1997). The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of the matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as to help identify members of gene families.
Step1: Select the program and database and paste or upload sequences
Step2: View the blast results and download the sequences you want
......

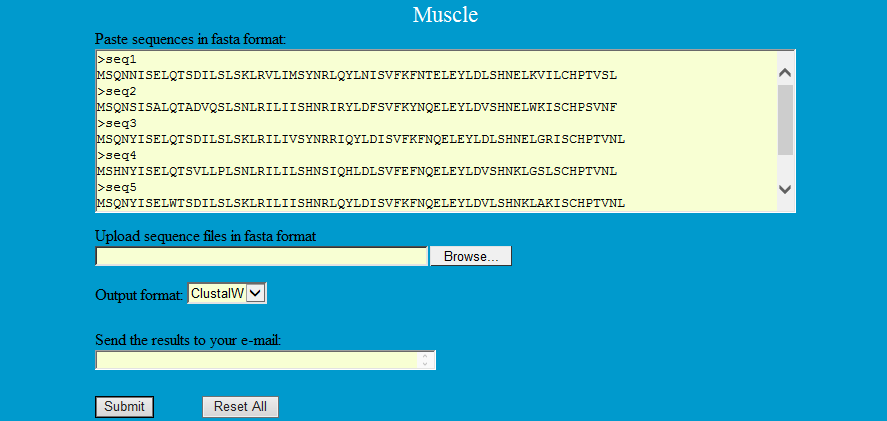
MUSCLE (Multiple Sequence Comparison by Log-Expectation), an accurate Multiple Sequence Alignment (MSA) tool, especially good with proteins, is claimed to achieve better average accuracy and better speed than ClustalW2 or T-Coffee, depending on the options (Edgar 2004).
Step1: Paste or upload the sequences in fasta format as follows and select the output format

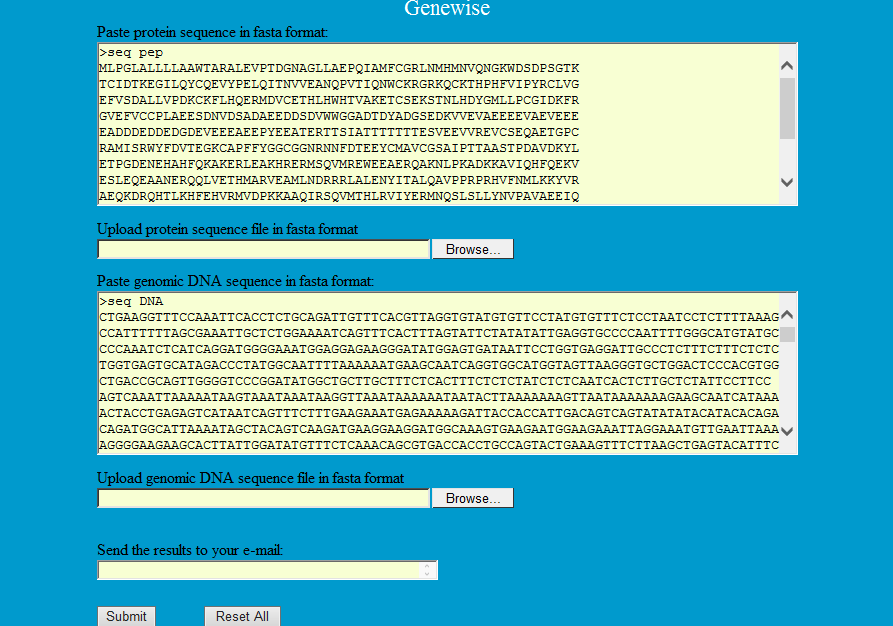
GeneWise, a Pairwise Sequence Alignment tool, compares a protein sequence to a genomic DNA sequence, allowing for introns and frames-shifting errors (Birney et al. 2004). GeneWise can provide highly accurate and sensitive predictions of gene structures.
Step1: Paste or upload the protein and corresponding genomic DNA sequences in fasta format as follows to predict gene structure

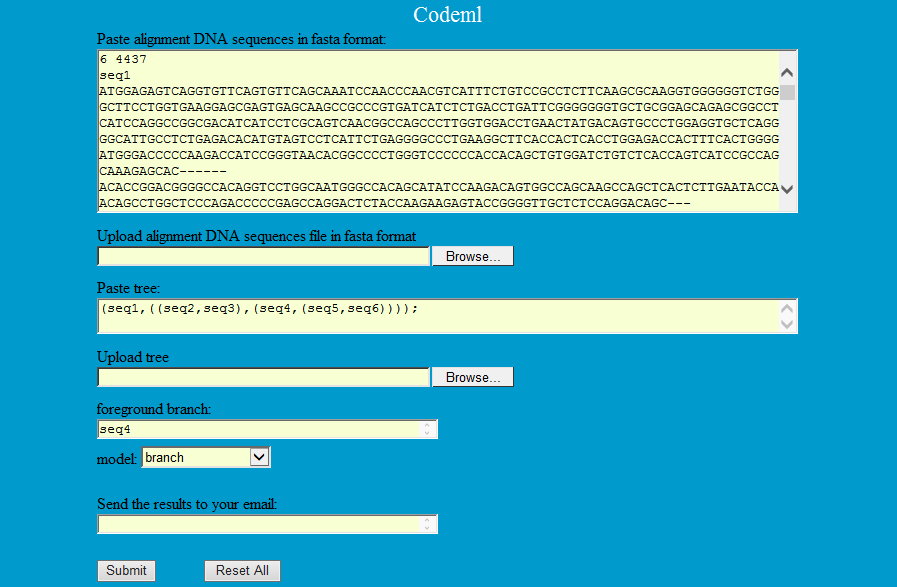
Codeml is a part of the PAML package, which is a suite of programs for phylogenetic analyses of DNA or protein sequences using maximum likelihood (ML) (Yang 2007).
Step1: Paste or upload alignment sequences of species they want to analysis with phylip format as follows and phylogenetic tree of these species, then select the model to finish the compute by codeml.
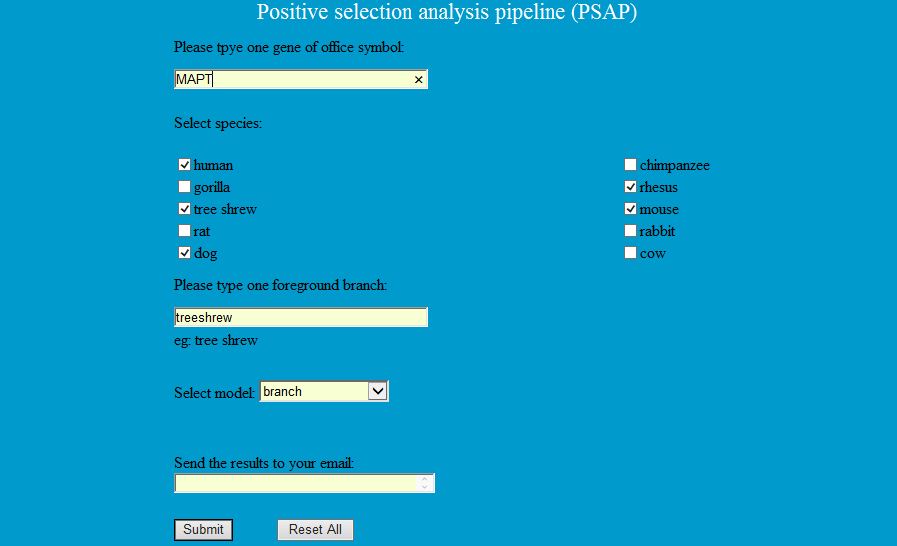
Positive selection analysis pipeline (PSAP) is developed based on perl script, muscle and codeml from PAML package (Yang 2007), which provides 10 species to preform positive selection analysis including human, chimpanzee, gorilla, rhesus monkey, Chinese tree shrew, mouse, rat, rabbit, dog and cow. When users type the gene, select the species and model, the pipeline will extract the related sequences according to ortholog relationship list and choose the phylogenetic tree from the phylogenetic tree list. Then the extracted CDS sequences will be aligned by Muscle 3.7 with the guidance of aligned protein sequences and finish positive selection analysis by codeml from PAML package.
Step1: Type the gene symbol and foreground branch, select species and model to finish the compute by codeml.
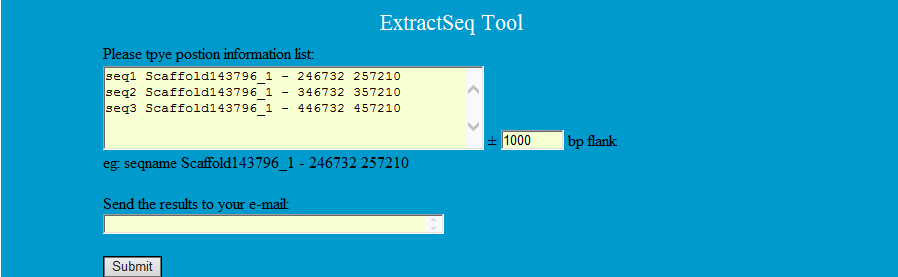
ExtractSeq can extract the sequence from Chinese tree shrew genome by scaffold position information
Step1: Type the position informations as follows to extract the corresponding sequences.
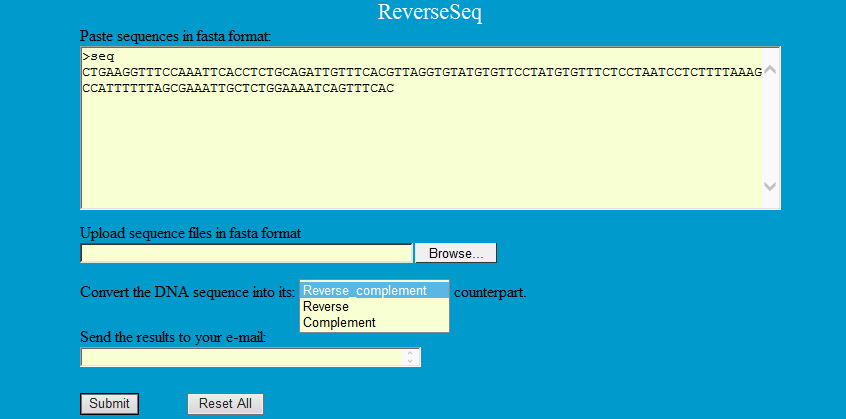
ReverseSeq can convert a DNA sequence into its reverse, complement, or reverse-complement counterpart. You may want to work with the reverse-complement of a sequence if it contains an ORF on the reverse strand.
Step1: Paste sequences in fasta format as follows to convert into its reverse, complement, or reverse-complement counterpart.
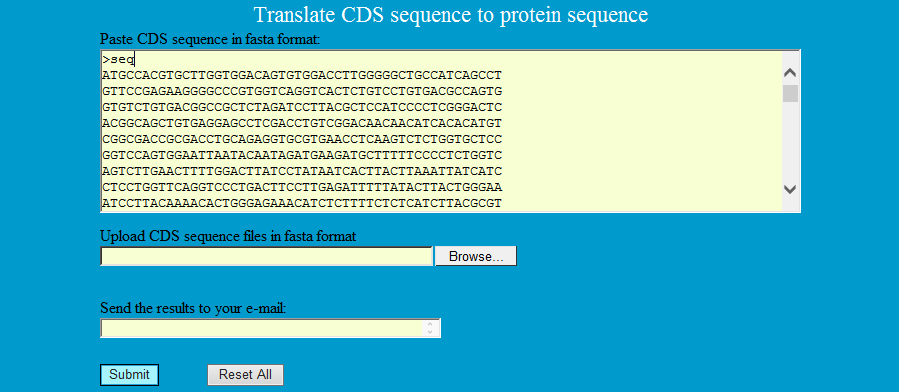
ReverseSeq can translate coding DNA sequence to protein sequence.
Step1: Paste CDS sequence in fasta format as follows to translate the corresponding protein sequence
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res, 25(17): 3389-3402.
Birney E, Clamp M, Durbin R. 2004. GeneWise and Genomewise. Genome Res, 14(5): 988-995.
Donlin MJ. 2007. Using the Generic Genome Browser (GBrowse). Curr Protoc Bioinformatics, Chapter 9: Unit 9 9.
Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res, 32(5): 1792-1797.
Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol, 24(8): 1586-1591.